Better data, better experiment, better science..!
Effective translational research requires effective and robust multi-omics data integration, in order to ensure complete and correct description of the mechanisms of complex disease such as cancer. This is very much about ‘the sample’ as we have to analyse it using different workflows and sampling methodologies. For example, a next gen sequencing workflow that obtains sequence data is very different to an LC-MS metabolomics workflow obtaining metabolite data. Bottom line: don’t forget about sample prep!
We are now well into the 21st Century and have already seen many truly pivotal advances in the life sciences. Our understanding of disease, at the molecular level, has benefitted exponentially. Rather, as we once mapped the world’s oceans, we continue to construct ever more detailed ‘maps’ of disease. As these maps become more detailed, we see ‘pathways’ that describe the mechanism of specific diseases, be it neurodegenerative, cancer, cardiovascular or other. As a consequence, we have developed complex methods for culturing cells and reconstructed tissues, through measurements of cellular changes at a molecular level (so-called ‘omics’ technologies) to vastly improved computing capacity that can be applied to make sense of the huge volumes of data generated from multiple omics approaches (e.g., genomics, transcriptomics, proteomics, and metabolomics), as is the motivation of the human microbiome project.
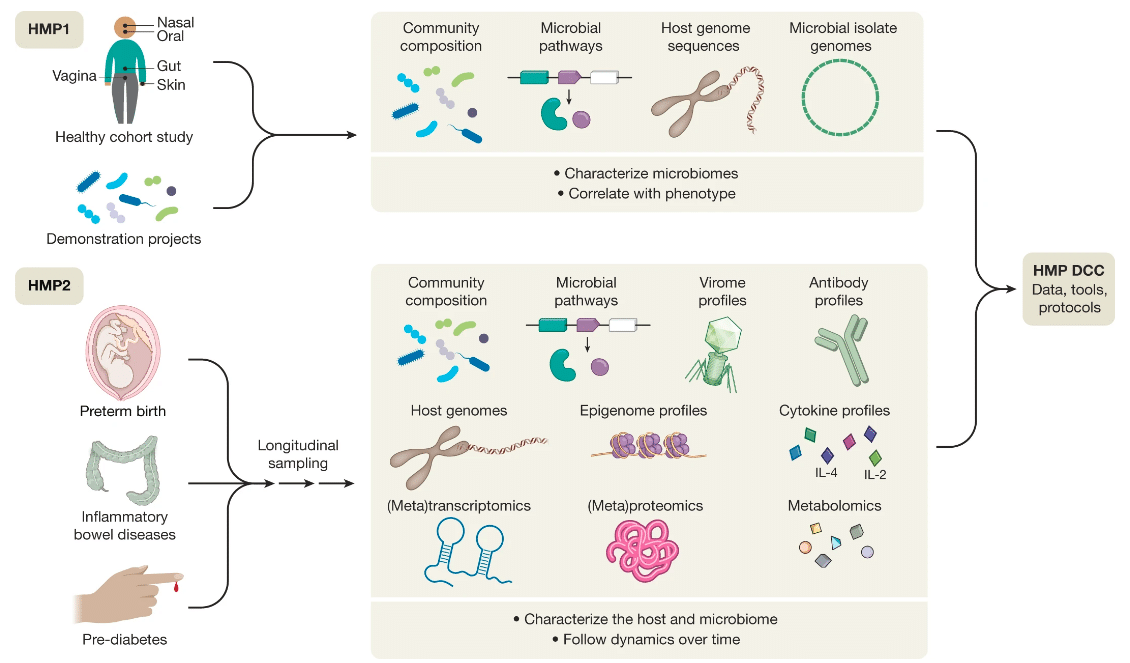
Multi-omics data integration for the human microbiome project
High-quality multi-omics studies require:
- proper experimental design
- thoughtful selection, preparation, and storage of appropriate biological samples
- careful collection of quantitative multi-omics data and associated meta-data
- better tools for integration and interpretation of the data,
- agreed minimum standards for multi-omics methods and meta-data,
Ideally such a study should involve multi-omics data being generated from the same sample(s) though this can be more challenging due to limitations in sample access, biomass, and cost. A good example of this is the fact that formalin-fixed paraffin-embedded (FFPE) tissues are compatible with genomic studies but not with transcriptomic or, until recently, proteomic studies, due to the fact that formalin does not stop RNA degradation and paraffin can interfere with MS performance thus affecting both proteomic and metabolomic assays.
There is a case to be made for basing multi-omics experimental design requirements on that used for metabolomics. Metabolomics experiments are highly compatible with a wide range of biological samples including blood, serum, plasma, cells, cell culture and tissues; which also happen to be preferred for transcriptomic, genomic and proteomic studies; assuming a consistent sample storage protocol.
Metabolomics experiments require fast sample processing times, demanding the same of transcriptomic and proteomic studies, in order to ensure consistency. Moreover, metabolites are also particularly sensitive to environmental influences (diurnal cycles, heat, humidity, diet, age, developmental stage and social interactions) and the tracking of sample meta-data is very important to mitigate the effects of environmental confounders, and to facilitate transparency and reproducibility.
It is ”all about the sample”.. automating sample prep for multi-omics integration minimises error.
As stated at the start, it is ‘all about the sample’ and the integration of omics data strongly depends on rigorous and consistent sample prep, to reduce sample variation over pretty short periods of time. This demands accurate tracking of each step of the sample prep conducted in the different omics workflows, including the multitude of basic, yet critical liquid handling steps, involving anything from serial dilutions to plate normalization.
Robust, repeatable sample prep, requires rigorous adherence to protocol in order to obviate the risk of contamination, ease of method transfer, and most important, the ability to digitally capture a complete and accurate record of every single step of the sample preparation, including labware used, pipette calibration data, reagent prep temperatures, tube or microplate agitation speeds, and much more, in order to facilitate the translation of omics data sets upon completion of each analytical workflow.
Andrew Alliance’s cloud-native OneLab ecosystem ensures rapid, intuitive and precise protocol creation, with ease of method transfer to other labs, minimizing intra- or inter-lab variability.

Andrew+ offers fully automated pipetting, as well as more complex manipulations, using a wide range of Domino Accessories and Andrew Alliance electronic pipettes. It executes OneLab protocols, enabling rapid transition from laborious manual procedures to error-free, robotic workflows.
Wireless execution of these protocols on its increasing range of connected devices, enabling fully automated liquid handling (Andrew+), guided pipetting (Pipette+), shaking (Shaker+), rapid heating/cooling (Peltier+), magnetic bead separation (Magnet+), or micro-elution for SPE (Vacuum+) defines not only the blueprint for the connected lab of the future but also the capability necessary to realise the demanding sample prep requirements of multi-omics data integration, and therefore, pathway-based translational research
Learn more at www.andrewalliance.com
